









| 目的 | 慢性便秘症患者の便意と糞便量、直腸感覚閾値と排便に対するグーフィス®(エロビキシバット)の影響について調べること。 |
| 試験デザイン | 前向き無作為化並行群間二重盲検プラセボ対照比較試験 |
| 対象 | 第1回目の来院時(Visit 1)に、文書同意を得た患者に血液検査、問診を行い、適格性が確認できた患者を仮登録し、排便日誌の記録を依頼した。1週間の観察期間の後、3日間の準備期間を設け、準備期間の最終日にホテルに宿泊した参加者を来院させて症状を記録し、正式登録した(Visit2)。身体検査、直腸感覚閾値検査を実施し、参加者をプラセボ投与群(以下プラセボ群 n= 8)とグーフィス®10 mg/日投与群(以下グーフィス®群n= 9)に無作為に割り付け、試験薬を1週間投与した。 試験薬投与中のビサコジル坐剤の服用を、条件に従い1回もしくは2回許可した。 参加者は、試験薬投与中の7日間のうち、1日目~5日目までは通常通り自宅で過ごし、試験日前日の6日目はホテルに宿泊し指定の食事を摂った。7日目に来院し、身体検査、血液検査、直腸感覚閾値の検査を受けた(Visit 3)。 試験プロトコール
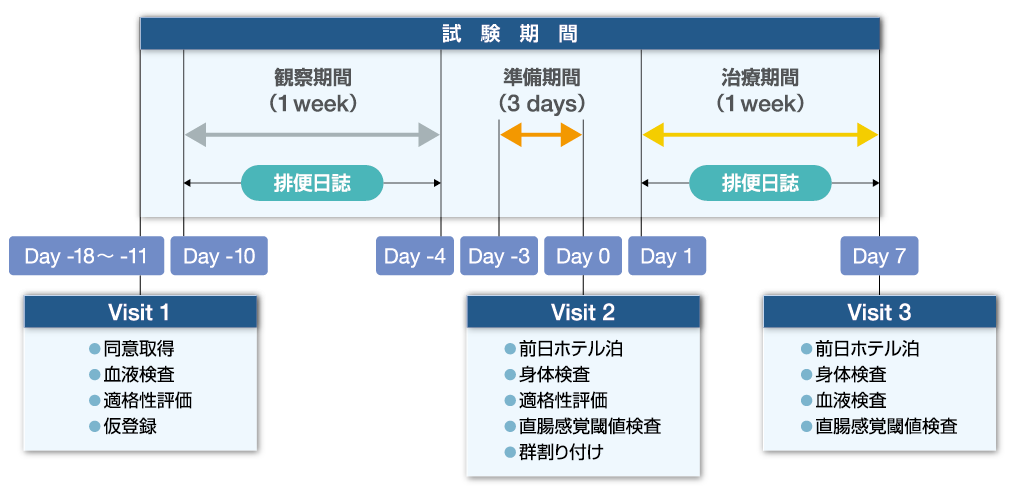 Manabe N, et al.: BMJ Open Gastroenterol 2023; 10: e001257 より改変 Manabe N, et al.: BMJ Open Gastroenterol 2023; 10: e001257 より改変直腸感覚閾値検査について 直腸感覚閾値の検査は、Visit 2、Visit 3において行われた。 Visit 3での検査は、試験薬投与後5時間までに行われた。検査前日の夕食、検査当日の朝食には既定の食事を摂取させた(夕食:623kcal<蛋白質17.4g 脂質 22.8g>、朝食:682kcal<蛋白質 27.4g 脂質22.7g>)。検査の4時間以上前から摂食を禁止、水のみ摂取可とした。 バロスタット法により、FCSV(first constant sensation volume:直腸の伸展を感じる容量)、DDV(defaecatory desire volume:便意を感じる容量)、MTV(maximum tolerable volume:最大許容容量)の3つの感覚閾値を5分間隔で測定した。測定値の変動が大きい場合は1回または2回追加測定を行い、中央値を算出した。 |
| 評価項目 | 【主要評価項目】慢性便秘症患者のDDVに対するグーフィス®の影響 【副次評価項目】慢性便秘症患者のFCSV、MTV及び排便に対するグーフィス®の影響 |
| 解析計画 |
|
| Limitation |
|








監修者コメント
本調査では、日常診療下のグーフィス®の安全性と有効性を評価した。その結果、ADR発現率は、4週間投与試験6.35%、52週間投与試験5.40%、有効性については、4週間投与試験、52週間投与試験ともに週当たりの排便回数がグーフィス®投与後に増加したなどの結果が得られた。また、グーフィス®投与後の排便までの時間は平均約6時間であり、ICC(級内相関係数)に基づく検討から、排便がほぼ一貫した時間で起きていたと考えられる。
本調査の結果を踏まえ、冒頭でも紹介した、計画排便のためのより積極的な介入として、グーフィス®を用いて排便時間を予測できる治療が行えれば、患者のアドヒアランスや医師の負担軽減の観点からも有効ではないかと考える。以下にグーフィス®を用いた計画排便の実践例を示す。
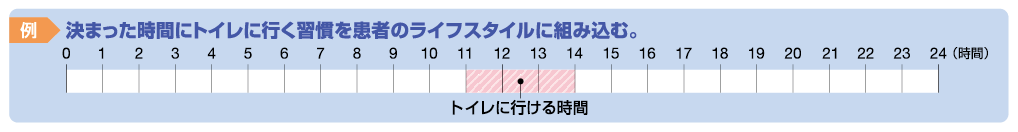 トイレに行ける時間の6時間前にグーフィス®を服用する。
トイレに行ける時間の6時間前にグーフィス®を服用する。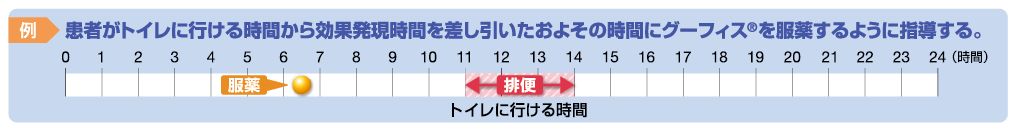 監修:中島 淳先生
監修:中島 淳先生




















